设计方案的制定:R&D部根据批准的设计开发任务书制定设计开发计划。它包括内容设计和开发的所有阶段(计划阶段、输入阶段、输出阶段、审查阶段、试验阶段、试验阶段、定稿阶段等。),适用于各阶段的设计评审、验证和确认活动,各阶段的任务、责任人、进度要求以及需要增加和调整的资源。设计方案经R&D部门审核后,提交总经理审批。设计输入的评审:设计输入完成后,R&D部组织相关部门和人员对设计输入进行评审。评审内容包括设计和开发任务书规定内容的完整性和合理性,产品的预期用途、功能和结构,满足顾客要求的程度,国家法律法规的要求以及设计和开发计划所包含内容和资源的调整。R&D部根据评审情况编写设计输入评审报告,包括需要采取的措施。R&D部门负责人审核后,该报告将提交给总经理审批,R&D部门将跟踪需要采取的措施。自2018年医疗采购器械要求提出优先考虑国产器械后,国内医疗器械开发呈井喷式增长!新型医疗器械
风险管理职责和权限分配:a)技术部总经理/经理:应是总负责人,风险管理活动的批准者,提供资源以确保人员的质量来完成风险管理活动;b)研发工程师:负责设计和开发过程中的风险管理活动,对风险分析、风险评估、风险控制措施的实施和验证以及剩余风险评估进行相关记录并编写风险管理报告。c)质量部,负责整理和控制体系文件,收集和反馈产品实现过程中已知和可预见的危害,以及生产和生产后的信息给R&D部。d)生产部、销售部、临床注册部:收集产品实现过程中已知和可预见的危害,以及生产和生产后的信息,并反馈给R&D部。e)R&D部门和风险管理活动小组:定期审查风险管理活动,以确保其有效性和真实性。医疗器械设计属于什么专业医疗器械设计开发主要包括哪些部分?
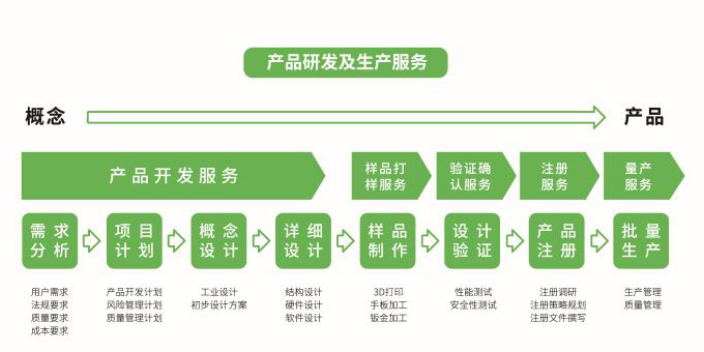
设计验证的评估设计验证的评审方法分为内部评审和外部评审。内部鉴定:R&D部将组织相关部门和人员召开产品鉴定会,检查产品是否合格。召开评审会议时,R&D部准备评审资料,包括设计和开发计划、输入文件、技术文件、评审和验证记录、产品试制报告、顾客验证等。,并在此基础上得出评估结论。鉴定包括产品满足设计任务书和顾客要求的评价;产品图纸、设计文件和工艺文件是否完整、规范,能否正确指导生产评价;对产品结构、功能、工艺、技术指标、先进生产能力、用户的可靠性、安全性和稳定性以及采用国内外先进技术标准的评价;满足顾客要求的程度以及符合国家法律法规和国家标准的情况;是否具备产品定型的条件。对外,R&D部将确定的产品送国家认可的第三方医疗器械检验机构进行验证,或送有资质的医院进行临床试验。验证成功后才能进行临床确认。没有经过验证的新产品安全性得不到保证,所以不允许进入临床。R&D部将**终确定的产品送至国家认可的医疗器械检测机构进行型式试验、可靠性试验和认证试验,并根据确定的**终意见整理出产品符合性报告,报总经理批准。
设计应该贯穿于产品设计、开发、生产的每一个阶段。例如,在可行性研究/原型阶段、产品开发阶段、临床试验开始前、临床试验期间和生产开始后、产品变更阶段。风险管理和分析是设计控制的重要组成部分和要求。风险管理是分析、评估、控制和缓解产品质量风险的系统过程,贯穿于整个产品生命周期,包括产品发生变化时的风险评估和控制。制造商应明确与产品相关的危害并使相关风险小化,判断和评估相关风险,控制这些风险,并监视控制措施的有效性。由于人的因素在产品开发和使用过程中无处不在,因此在设计控制中考虑人的因素和产品风险控制是非常重要的。ISO 1348 5中对医疗器械设计开发的相关要求和说明。

设计和开发输入的范围取决于仪器的复杂程度和风险,一般应包括功能、性能、安全和监督的要求。要求的内容应在设计输入要求中用工程术语明确说明。应在设计和开发输入中投入足够的时间和资源,以保证输入的完整性和各方位覆盖,输入阶段占项目总时间的30%以上。在验证过程中,不可避免地需要更改设计输入,应控制设计输入要求的更改。纠正问题的设计更改可能会导致必须解决的新问题。设计和开发全过程的变更应评价对产品实现的总体影响。医疗器械产品的设计开发过程是需要进行质量控制的重要环节,无论是标准还是法规都对产品的设计开发有要求。浙江医疗器械设计开发诚信推荐
医疗器械设计的细节直接影响到患者的健康和生命,事实上,医械的设计和开发总是存在一定程度的风险。新型医疗器械
风险管理计划应包括以下条款:a)计划风险管理活动的范围,确定和描述适用于计划每个要素的医疗器械和生命周期阶段(设计-验证-设计转化-生产-生产后管理)(时间阶段因素);b)职责和权力的分配(人的因素);c)评审风险管理活动的要求(风险审计);d)在标准风险可接受的基础上,制造商制定用于确定可接受策略的风险,包括标准风险可接受时的发生概率和危害估计(风险接受的标准和评估标准);e)根据制造商制定的可接受风险政策评估总体剩余风险的方法和总体剩余风险的可接受标准(剩余风险的评估和评价标准);f)验证风险控制措施的实施和有效性的活动(风险控制措施的验证);g)与收集和审查生产和后期生产信息有关的活动(生产和生产后风险控制)。新型医疗器械
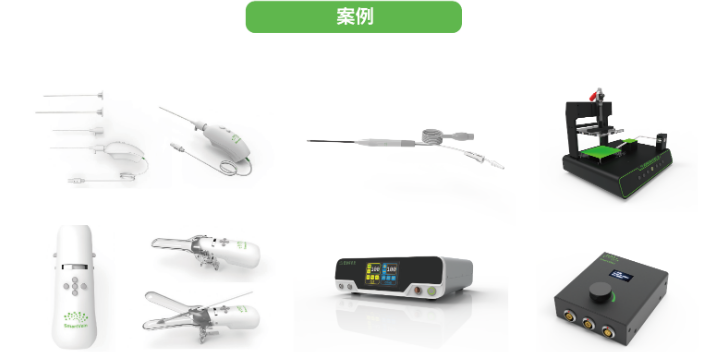
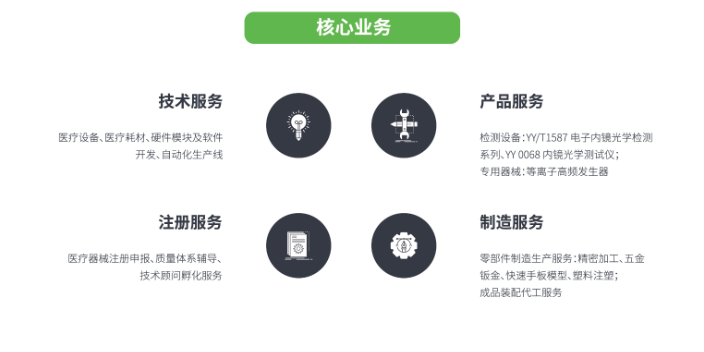
思脉得(嘉兴)医疗科技有限公司是以提供技术服务,注册服务,产品服务,制造服务为主的有限责任公司(自然),思脉得医疗集团是我国医药健康技术的研究和标准制定的重要参与者和贡献者。思脉得医疗集团以技术服务,注册服务,产品服务,制造服务为主业,服务于医药健康等领域,为全国客户提供先进技术服务,注册服务,产品服务,制造服务。将凭借高精尖的系列产品与解决方案,加速推进全国医药健康产品竞争力的发展。